


Klinische Forschungsgruppe KFO 5029
Präzisionsmedizin bei Erkrankungen mit früh-manifester Reduktion der Knochenmineraldichte
Erniedrigte Knochenmineraldichte wird oft in der älteren Bevölkerungsgruppe diagnostiziert, kann aber auch vor dem Alter von 50 Jahren auftreten, was oft mit Frakturen und einer verminderten Lebensqualität verbunden ist. In vielen Personen mit einer früh einsetzenden Erniedrigung der Knochenmineraldichte wurden Mutationen in spezifischen Genen identifiziert, welche wichtig für die Funktion von Knochenzellen sind. Unsere klinische Forschungsgruppe (ProBone) hat zum Ziel, die Ursachen der früh einsetzenden Erniedrigung der Knochenmineraldichte für einzelne Patient:innen herauszufinden, um die bestmögliche personalisierte Therapie zu etablieren. Wir werden zudem molekulare Studien durchführen, um die Funktion spezifischer Gene für das Skelett zu verstehen, um in Zukunft neue Therapien zu ermöglichen.
Sprecher: Prof. Dr. Michael Amling
Leiter: Prof. Dr. Ralf Oheim
Wissen und Forschen - ProBone
Eine kurze Vorstellung des Hintergrundes und Konzeptes von ProBone im Rahmen der UKE "Wissen und Forschen" Serie.
-
Präzisionsmedizin bei Erkrankungen mit früh-manifester Reduktion der Knochenmineraldichte
Die beantragte klinische Forschungsgruppe hat das Ziel, Präzisionsmedizin zur Diagnose und personalisierten Therapie von Erkrankungen mit früh-manifester Reduktion der Knochenmineraldichte (KMD) zu implementieren. Die Gemeinsamkeiten in dieser heterogenen Gruppe sind niedrige KMD, begleitet von nicht-traumatischen / inadäquaten Frakturen, vor dem 50. Lebensjahr nach Ausschluss bekannter sekundärer Ursachen. Aufgrund des hohen Leidensdrucks und der reduzierten Lebensqualität ist das Verständnis der zellulären und molekularen Grundlagen der entsprechenden Pathologien essentiell, um spezifische Therapien zu optimieren oder zu etablieren. Zur Zeit erhalten diese Patient:innen oft die unspezifische Diagnose “idiopathische Osteoporose“, und es gibt keine therapeutischen Richtlinien, wie dem KMD-Verlust entgegen zu wirken ist und/oder weitere Frakturen zu verhindern sind. Da ein Begriff wie “idiopathische Osteoporose“ letztendlich nur die bestehende Wissenslücke aufzeigen kann, ist unsere zentrale Hypothese, dass durch Präzisionsmedizin in vielen Fällen eine eindeutige genetische oder nicht-genetische Ursache der Erkrankung identifiziert und ggf. zielgereichtet behandelt werden kann.
Unser Vorhaben basiert auf Erkenntnissen aus Untersuchungen von Patient:innen, die bei der Spezialambulanz für seltene muskuloskelettale Erkrankungen (National Bone Board) am Institut für Osteologie und Biomechanik (IOBM) vorstellig wurden. Aufgrund der hohen Zahl von Patient:innen am IOBM (ca. 10.000 pro Jahr), hat das National Bone Board seit 2015 klinische und genetische Daten von über 1.000 Patient:innen mit frühzeitig erniedrigter KMD dokumentiert. Da sich diese Zahl voraussichtlich um ca. 100 Fälle pro Jahr erhöhen wird, möchten wir, basierend auf dieser weltweit einzigartigen und außergewöhnlich großen Kohorte, fundierte Analysen durchführen, um das molekulare Verständnis dieser Erkrankungen substantiell zu verbessern. Gemeinsam mit 9 verschiedenen Instituten/Kliniken des Universitätsklinikums Hamburg-Eppendorf und der Universität Hamburg wird das multidisziplinäre, translationale Forschungsprogramm, unter Einsatz neuester Technologien, ein ganzheitliches klinisches und molekulares Verständnis dieser Krankheitsentität ermöglichen.
In der ersten Förderperiode verfolgen wir verschiedene explorative Strategien, gestützt durch bioinformatische Analysen, um folgende Ziele zu erreichen: i) Stratifizierung der Kohorte und personalisierte Therapie, ii) Molekulares Verständnis Patient:innen-spezifischer zellulärer Störungen, iii) Identifizierung bisher unbekannter Krankheits-assoziierter Mechanismen. In einer zweiten Förderperiode würden wir unsere Forschung im Hinblick auf zuvor unbekannte Pathologien intensivieren sowie die erzielten Erkenntnisse nutzen, um neue innovative Therapie-Optionen zu entwickeln. Diese könnten zudem für die Behandlung Alters-assoziierter Skelett-Erkrankungen, wie der Osteoporose, relevant sein, welche ein ständig wachsendes Problem in der Bevölkerung darstellen. -
Projekt 1: Krankheitsstratifizierung, Behandlungsüberwachung und Etablierung von Modellsystemen

Projekt 1 hat zum Ziel, Patient:innen zu identifizieren und zu charakterisieren, Proben, Daten und Wissen anderen Projekten zur Verfügung zu stellen, sowie den Einfluss von Therapien, wenn eingeleitet, zu dokumentieren. Projekt 1 ist auch für die Generierung Krankheits-spezifischer Modellsysteme zuständig, um den Einfluss pathogener Varianten in neuen und/oder wenig verstandenen Gene zu untersuchen.
-
-
Projekt 2: Klinische Charakterisierung angeborener Erkrankungen mit früh-manifester Reduktion der Knochenmineraldichte vom Säuglings- bis zum Jugendalter
Das Hauptziel von Projekt 2 ist die Identifizierung und umfassende Charakterisierung skelettaler Manifestationen von sich im Kindesalter manifestierenden Erkrankungen mit niedriger Knochendichte sowie die Datenerhebung und -analyse im Rahmen einer neu entwickelten Datenbank. Zusätzlich streben wir die Entwicklung eines experimentellen Behandlungsansatzes für die häufigste genetische Ursache der Mukolipidose Typ II (MLII) an.
-
-
Projekt 3: Identifizierung neuer Krankheitsgene für monogene Formen von Erkrankungen mit früh-manifester Reduktion der Knochenmineraldichte
Projekt 3 nutzt Hochdurchsatzsequenziertechnologien zur Aufdeckung neuer Krankheitsgene für eine primäre, früh-manifeste Reduktion der Knochenmineraldichte. Für jedes neu identifizierte monogene Krankheitsgen werden spezifische Kombinationen von klinischen Merkmalen, osteologischen Parametern, immunologischen und/oder metabolischen Veränderungen bei den betroffenen Personen herausgearbeitet. Funktionelle Studien dienen zur Bestätigung der Pathogenität der genetischen Varianten und zur Untersuchung des Pathomechanismus.
-
-
Projekt 4: Molekulare Grundlagen nicht-klassischer Formen der Osteogenesis imperfecta
Im Fokus von Projekt 4 sind die nicht-klassischen Formen der Osteogenesis imperfecta, die durch geringe Knochenmasse und skelettale Fragilität charakterisiert sind. Wir wollen die dynamischen molekularen Prozesse während der Knochenbildung besser verstehen, als auch Schlüsselmoleküle und Signalwege unter pathophysiologischen Bedingungen identifizieren, um kausale Therapien etablieren zu können.
-
-
Projekt 5: Identifizierung genetischer und molekularer Ursachen der Hypophosphatasie
Projekt 5 befasst sich mit der Untersuchung der Hypophosphatasie, einer genetisch bedingten Erkrankung, die unter Anderem zu Mineralisationsstörungen und reduzierter Knochenmasse führt. Es sollen Genotyp-Phänotyp Korrelationen, neuartige Modellsysteme und therapeutische Ansätze sowie bisher unbekannte Pathomechanismen untersucht werden.
-
-
Projekt 6: Molekulare Analyse der bidirektionalen Wechselwirkung zwischen Knochen- und Lipidstoffwechsel
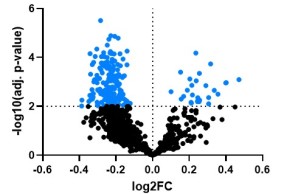
Im Projekt 6 sollen Metabolite identifziert werden, die von Osteoblasten freigesetzt werden und als Signalmoleküle möglicherweise den Knochen und systemischen Stoffwechsel regulieren. Die Relevanz und Spezifität der Veränderungen soll in genetischen Modellen untersucht werden, um neue therapeutische Optionen zur Behandlung von Patienten mit niedriger Knochenmineraldichte zu erforschen.
-
-
Projekt 7: Untersuchung des Zusammenspiels zwischen Knochen und Immunsystem bei früh manifestierenden KMD-Störungen
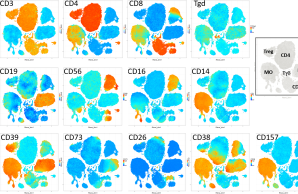
Eine umfassende Profilierung des Immunsystems bei Patienten mit früh einsetzender niedriger BMD wurde bisher nicht durchgeführt. In P7 wollen wir den Beitrag von Störungen des Immunsystems zu einer früh einsetzenden niedrigen Knochendichte sowie den Einfluss knochenbedingter Gendefekte und einer knochenkrankheitsspezifischen Behandlung auf die Funktion von Immunzellen untersuchen.
-
-
Projekt 8: Molekulares Verständnis der Frakturheilung bei Erkrankungen mit früh-manifester Reduktion der Knochenmineraldichte
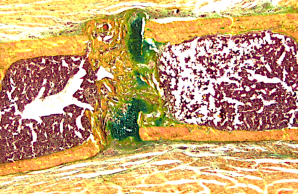
In Projekt 8 wird untersucht, inwieweit spezifische Genvarianten den mehrstufigen Prozess der Frakturheilung auf zellulärer und molekularer Ebene beeinflussen. Neben genetischen, metabolischen und immunologischen Aspekten wird auch die Eignung spezifischer pharmakologischer Interventionen zur Verbesserung der Knochenregeneration analysiert.
-
-
Projekt 9: Auswirkungen spezifischer Genvarianten auf die Wachstumsfuge und den Gelenkknorpel bei Erkrankungen mit früh-manifester Reduktion der Knochenmineraldichte
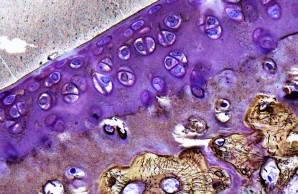
Projekt 9 umfasst die Charakterisierung von Patienten, die neben einer früh manifesten Reduktion der Knochenmineraldichte auch eine Skelettdysplasie und/oder Arthrose aufweisen. Zur Untersuchung von Pathomechanismen werden histomorphometrische und molekulare Analysen sowie Krankheitsmodelle unter Verwendung von pluripotenten Stammzellen und Mausmodellen eingesetzt.
-
-
Zentralprojekt 1: Herstellung humaner induzierter pluripotenter Stammzellen
CP1 wird humane in vitro-Krankheitsmodelle entwickeln, um das Verständnis der Mechanismen einer früh-manifesten Reduktion der Knochenmineraldichte zu verbessern
-
-
Zentralprojekt 2: Bioinformatische Datenintegration und Multi-Omic Signalweg Analyse
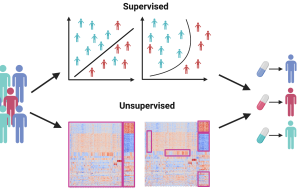
CP2 wird bioinformatische Unterstützung für jede Phase der computergestützten Analyse von klinischen und Omics-Daten bieten, die im Rahmen des ProBone-Projekts generiert werden. Dies umfasst die Sammlung von Proben, die Versuchsplanung, die harmonisierte Datenanalyse sowie die Erstellung und Validierung von Hypothesen. CP2 wird sich auf die Identifizierung von Patientensubtypen bei früh auftretenden Erkrankungen mit niedriger Knochenmineraldichte und die Krankheitsmechanismen konzentrieren, die diese Subtypen voneinander unterscheiden.
-