Arbeitsgruppe "Skelettale Pathobiochemie"
Leitung:
Prof. Dr. rer. nat. Sandra Pohl
Heisenberg-Professorin für Subzelluläre Osteologie
Telefon:
Mail:
s.pohl(at)uke.de
- Forschung
- Team
- Publikationen
- Förderung
- Stellenangebote
-
Forschung
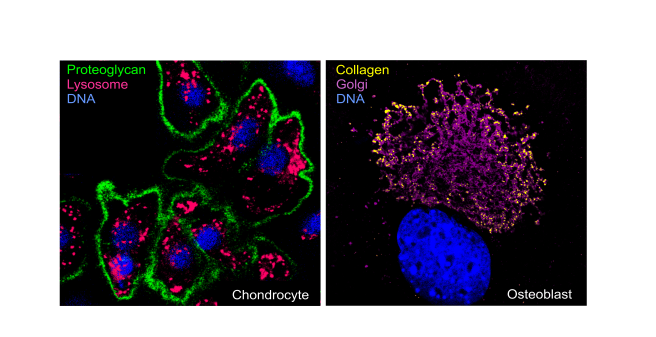
Die organische skelettale Matrix besteht aus strukturell sehr verschiedenen Makromolekülen wie Collagene, Proteoglykane und Glykoproteine. Diese Proteine werden nach Synthese und posttranslationalen Modifikationen im endoplasmatischen Reticulum und im Golgi-Apparat von Chondrocyten im Knorpel und Osteoblasten im Knochen sezerniert. Dagegen erfolgt der Abbau der skelettalen Matrixproteine über hydrolytische Enzyme wie z. B. lysosomale Enzyme von Zellen der extrazellulären Matrix. Für die Entwicklung aber auch den Erhalt des Skeletts muss die hochdynamische skelettale Matrix des Knochens und des Knorpels kontinuierlich umgebaut werden.
Pathophysiologie und molekulare Ursachen von Skeletterkrankungen im Kindesalter
Gestörte Stoffwechselvorgänge in Knochen- und Knorpelgewebe führen zu einer Vielzahl von Erkrankungen im Kindesalter, zu denen auch die seltenen genetisch-bedingten lysosomalen Speicherkrankheiten und Osteogenesis imperfecta gehören. Biochemisch sind diese Krankheiten durch Defizienz oder Inaktivität von spezifischen Proteinen charakterisiert, die eine Vielzahl von molekularen Stoffwechselwegen im Knochen und Knorpel koordinieren. Dies manifestiert sich in den betroffenen Patienten in einer breiten Heterogenität der skelettalen Anomalitäten.Unsere Forschungsgruppe möchte die molekularen Mechanismen verstehen, die die Synthese und den Abbau der skelettalen Matrix im Gleichgewicht halten, und die pathophysiologischen Ursachen genetisch-bedingter Skeletterkrankungen im Kindesalter aufklären. Dafür untersuchen wir Mausmodelle der Erkrankungen, Zellkultursysteme und Patientenmaterial unter Verwendung einer Kombination aus biochemischen, molekular- und zellbiologischen aber auch histologischen und immunhistochemischen Methoden in engen Kooperationen mit verschiedenen Forschungsgruppen innerhalb und außerhalb des IOBM.
-
-
Team
 Prof. Dr. rer. nat.Sandra Pohl
Prof. Dr. rer. nat.Sandra Pohl- Forschungsgruppenleiterin
Standort
Campus Forschung I - N27 , 1. Etage, Raumnummer 01.006 Maike BuckM. Sc.
Maike BuckM. Sc.- Doktorandin
Standort
Campus Forschung I - N27 , EG, Raumnummer 0.007 Kenneth SchöneckM. Sc.
Kenneth SchöneckM. Sc.- Doktorand
Standort
Campus Forschung I - N27 , EG, Raumnummer 0.007 Standort
Standort
Campus Forschung I - N27 , 1. Etage, Raumnummer 01.008 Standort
Standort
Campus Forschung I - N27 , 1. Etage, Raumnummer 01.008.1 Naemi Euchner
Naemi Euchner- Medizin-Doktorandin
Standort
Campus Forschung I - N27 , 1. Etage Judith Zeidler
Judith Zeidler- Medizin-Doktorandin
Standort
Campus Forschung I - N27 , 1. Etage
-
Publikationen
2025
Absolute quantification of lysosomal proteins by multiple reaction monitoring mass spectrometry and QconCAT protein standards.
Mosen PR, Moharana B, Fajardo-Callejon S, Kaade E, Rösel N, Omidi M, Sakson R, Ruppert T, Pohl S, Gieselmann V, Winter D (2025) bioRxiv https://doi.org/10.1101/2025.01.09.632238 (preprint)2024
Inactivation of spermine synthase in mice causes osteopenia due to reduced osteoblast activity.
Yorgan TA, Zhu Y, Wiedemann P, Schöneck K, Pohl S, Schweizer M, Amling M, Barvencik F, Oheim R, Schinke T (2024) J Bone Miner Res 56. doi: 10.1093/jbmr/zjae156O-GlcNAcylation in the osteoblast lineage - boosting the complexity of Wnt-stimulated bone formation.
Pohl S, Schinke T (2024) EMBO Rep doi: 10.1038/s44319-024-00242-22023
Mice heterozygous for an osteogenesis imperfecta-linked MBTPS2 variant display a compromised subchondral osteocyte lacunocanalicular network associated with abnormal articular cartilage.
Danyukova T, Alimy AR, Velho RV, Yorgan TA, Di Lorenzo G, von Kroge S, Tidow H, Wiegert JS, Hermans-Borgmeyer I, Schinke T, Rolvien T, Pohl S (2023) Bone 177: 116927CNS manifestations in mucolipidosis type II - A retrospective analysis of longitudinal data on neurocognitive development and neuroimaging in eleven patients.
Ammer LS, Täuber K, Perez A, Dohrmann T, Denecke J, Santer R, Blümlein U, Ozga AK, Pohl S, Muschol NM (2023) J Clin Med 18: 4114Non-functional ubiquitin C-terminal hydrolase L1 drives podocyte injury through impairing proteasomes in autoimmune glomerulonephritis.
Reichelt J, Sachs W, Frömbling S, Fehlert J, Studencka-Turski M, Betz A, Loreth D, Blume L, Witt S, Pohl S, Brand J, Czesla M, Knop J, Florea BI, Zielinski S, Sachs M, Hoxha E, Hermans-Borgmeyer I, Zahner G, Wiech T, Krüger E, Meyer-Schwesinger C (2023) Nat Commun 14: 21142022
A homozygous hypomorphic BNIP1 variant causes an increase in autophagosomes and reduced autophagic flux and results in a spondylo-epiphyseal dysplasia.
Holling T, Bhavani GS, von Elsner L, Shah H, Kausthubham N, Bhattacharyya SS, Shukla A, Mortier GR, Schinke T, Danyukova T, Pohl S, Kutsche K, Girisha KM (2022) Hum Mutat doi: 10.1002/humu.24368Site-1 and site-2 proteases: A team of two in regulated proteolysis
Danyukova T, Schöneck K, Pohl S (2022) Biochim Biophys Acta Mol Cell Res 1869: 1191382021
Pathogenic variants in GNPTAB and GNPTG encoding distinct subunits of GlcNAc-1-phosphotransferase differentially impact bone resorption in patients with mucolipidosis type II and III
Di Lorenzo G, Westermann LM, Yorgan TA, Stürznickel J, Ludwig NF, Ammer LS, Baranowsky A, Ahmadi S, Pourbarkhordariesfandabadi E, Breyer SR, Board TN, Foster A, Mercer J, Tylee K, Velho RV, Schweizer M, Renné T, Braulke T, Randon DN, Sperb-Ludwig F, de Camargo Pinto LL, Moreno CA, Cavalcanti DP, Amling M, Kutsche K, Winter D, Muschol NM, Schwartz IVD, Rolvien T, Danyukova T, Schinke T, Pohl S (2021) Genet Med 23: 2369-2377Mucolipidosis type II and type III: a systematic review of 843 published cases
Dogterom EJ, Wagenmakers MAEM, Wilke M, Demirdas S, Muschol NM, Pohl S, Meijden JCV, Rizopoulos D, Ploeg ATV, Oussoren E (2021) Genet Med 23: 2047-2056Transgenic inhibition of interleukin-6 trans-signaling does not prevent skeletal pathologies in mucolipidosis type II mice
Westermann LM, Baranowsky A, Di Lorenzo G, Danyukova T, Soul J, Schwartz JM, Hendrickx G, Amling M, Rose-John S, Garbers C, Schinke T, Pohl S (2021) Sci Rep 11: 3556Is hematopoietic stem cell transplantation a therapeutic option for mucolipidosis type II?
Ammer LS, Pohl S, Breyer SR, Aries C, Denecke J, Perez A, Petzoldt M, Schrum J, Müller I, Muschol NM (2021) Mol Genet Metab Rep 26: 1007042020
Imbalanced cellular metabolism compromises cartilage homeostasis and joint function in a mouse model of mucolipidosis type III gamma
Westermann LM, Fleischhauer L, Vogel J, Jenei-Lanzl Z, Ludwig NF, Schau L, Morellini F, Baranowsky A, Yorgan TA, Di Lorenzo G, Schweizer M, de Souza Pinheiro B, Guarany NR, Sperb-Ludwig F, Visioli F, Oliveira Silva T, Soul J, Hendrickx G, Wiegert JS, Schwartz IVD, Clausen-Schaumann H, Zaucke F, Schinke T, Pohl S*, Danyukova T* (2020) Dis Model Mech 31: 1796-1814

Cover Image Westermann et al., Disease Models & Mechanisms Issue 31, 2020
Distinct modes of balancing glomerular cell proteostasis in mucolipidosis type II and III prevent proteinuria
Sachs W, Sachs M, Krüger E, Zielinsky S, Kretz O, Huber TB, Baranowsky A, Westermann LM, Velho RV, Ludwig NF, Yorgan TA, Di Lorenzo G, Kollmann K, Braulke T, Schwartz IV, Schinke T, Danyukova T, Pohl S*, Meyer-Schwesinger C* (2020) J Am Soc Nephrol 31: 1796-1814Hip morphology in mucolipidosis type II
Ammer LS, Oussoren E, Muschol NM, Pohl S, Rubio-Gozalbo ME, Santer R, Stuecker R, Vettorazzi E, Breyer SR (2020) J Clin Med 9: E728Enzyme replacement therapy in mice lacking arylsulfatase B targets bone-remodeling cells, but not chondrocytes
Hendrickx G, Danyukova T, Baranowsky A, Rolvien T, Angermann A, Schweizer M, Keller J, Schröder J, Meyer-Schwesinger C, Muschol N, Paganini C, Rossi A, Amling M, Pohl S*, Schinke T* (2020) Hum Mol Genet 29: 803-816Combined in-vitro and in-silico analyses of missense mutations in GNPTAB provide new insights into the molecular bases of mucolipidosis II and III alpha/beta
Danyukova T, Ludwig NF, Velho RV, Harms FL, Güneş N, Tidow H, Schwartz IV, Tüysüz B, Pohl S (2020) Hum Mutat 41: 133-1392019
Toward engineering the mannose 6-phosphate elaboration pathway in plants for enzyme replacement therapy of lysosomal storage disorders
Zeng Y, He X, Danyukova T, Pohl S, Kermode AR (2019) J Clin Med 8: E2190Mice deficient in the lysosomal enzyme palmitoyl-protein thioesterase 1 (PPT1) display a complex retinal phenotype
Atiskova Y, Bartsch S, Danyukova T, Becker E, Hagel C, Storch S, Bartsch U (2019) Sci Rep 9: 14185The lysosomal storage disorders mucolipidosis type II, type III alpha/beta and type III gamma: Update on GNPTAB and GNPTG mutations
Velho RV, Harms FL, Danyukova T, Ludwig NF, Friez MJ, Cathey SS, Filocamo M, Tappino B, Güneş N, Tüysüz B, Tylee KL, Brammeier KL, Heptinstall L, Oussoren E, van der Ploeg AT, Petersen C, Alves S, Saavedra GD, Schwartz IV, Muschol N, Kutsche K, Pohl S (2019) Hum Mutat 40: 842-864Humoral immune response in adult Brazilian patients with mucolipidosis III gamma
Sperb-Ludwig F, Alegra T, Velho RV, Ludwig N, Siebert M, Jobim M, Vairo F, Schwartz IVD (2019) Genet Mol Biol 42: 571-573A newly generated neuronal cell model of CLN7 disease reveals aberrant lysosome motility and impaired cell survival
von Kleist L, Ariunbat K, Braren I, Stauber T, Storch S, DanyukovaT (2019) Mol Genet Metab 126: 196-2052018
The lysosomal protein arylsulfatase B is a key enzyme involved in skeletal turnover
Pohl S, Angermann A, Jeschke A, Hendrickx G, Yorgan TA, Makrypidi-Fraune G, Steigert A, Kühn SC, Rolvien T, Schweizer M, Köhne T, Neven M, Winter O, Velho RV, Albers J, Streichert T, Pestka JM, Baldauf C, Breyer S, Stuecker R, Muschol N, Cox TM, Saftig P, Paganini C, Rossi A, Amling M, Braulke T, Schinke T (2018) J Bone Miner Res 33: 2186-2201
Cover Image Pohl et al., Journal of Bone and Mineral Research Issue 33, 2018
Lysosomal proteome and secretome analysis identifies missorted enzymes and their non-degraded substrates in mucolipidosis III mouse cells
Di Lorenzo G, Velho RV, Winter D, Thelen M, Ahmadi S, Schweizer M, De Pace R, Cornils K, Yorgan TA, Grüb S, Hermans-Borgmeyer I, Schinke T, Müller-Loennies S, Braulke T, Pohl S (2018) Mol Cell Proteomics 17: 1612-1626Loss of CLN7 results in depletion of soluble lysosomal proteins and impaired mTOR reactivation
Danyukova T, Ariunbat K, Thelen M, Brocke-Ahmadinejad N, Mole SE, Storch S (2018) Hum Mol Genet 27: 1711-17222017
GNPTAB missense mutations cause loss of GlcNAc-1-phosphotransferase activity in mucolipidosis type II through distinct mechanisms
Ludwig NF, Velho RV, Sperb-Ludwig F, Acosta AX, Ribeiro EM, Kim CA, Gandelman Horovitz DD, Boy R, Rodovalho-Doriqui MJ, Lourenço CM, Santos ES, Braulke T, Pohl S*, Schwartz IVD* (2017) Int J Biochem Cell Biol 92: 90-94Next-generation sequencing corroborates a probable de novo GNPTG variation previously detected by Sanger sequencing
Ludwig NF, Sperb-Ludwig F, Velho RV, Schwartz IVD (2017) Mol Genet Metab Rep 11: 92-99Site-1 protease and lysosomal homeostasis (Review)
Velho RV, De Pace R, Klünder S, Di Lorenzo G, Schweizer M, Braulke T, Pohl S (2017) Biochim Biophys Acta Mol Cell Res 1864: 2162-21682016
Identification of the interaction domains between α- and γ-subunits of GlcNAc-1-phosphotransferase
Velho RV, De Pace R, Tidow H, Braulke T, Pohl S (2016) FEBS Lett 590: 4287-4295Enigmatic in vivo GlcNAc-1-phosphotransferase (GNPTG) transcript correction to wild type in two mucolipidosis III gamma siblings homozygous for nonsense mutations
Velho RV, Ludwig NF, Alegra T, Sperb-Ludwig F, Guarany NR, Matte U, Schwartz IV. J Hum Genet 61: 555-5602015
Analyses of disease-related GNPTAB mutations define a novel GlcNAc1-phosphotransferase interaction domain and an alternative site-1 protease cleavage site
Velho RV, De Pace R, Klünder S, Sperb-Ludwig F, Lourenço CM, Schwartz IV, Braulke T, Pohl S (2015) Hum Mol Genet 24: 3497-3505Site-1 protease-activated formation of lysosomal targeting motifs is independent of the lipogenic transcription control
Klünder S, Heeren J, Markmann S, Santer R, Braulke T, Pohl S (2015) J Lipid Res 56: 1625-1632Subunit interactions of the disease-related hexameric GlcNAc-1-phosphotransferase complex
De Pace R, Velho RV, Encarnação M, Marschner K, Braulke T, Pohl S (2015) Hum Mol Genet 24: 6826-6835Biosynthesis, targeting, and processing of lysosomal proteins: pulse-chase labeling and immune precipitation
Pohl S, Hasilik A (2015) Methods Cell Biol 126: 63-832014
Mucolipidosis II-related mutations inhibit the exit from the endoplasmic reticulum and proteolytic cleavage of GlcNAc-1-phosphotransferase precursor protein (GNPTAB)
De Pace R, Coutinho MF, Koch-Nolte F, Haag F, Prata MJ, Alves S, Braulke T, Pohl S (2014) Hum Mutat 35:368-3762012
A novel mannose 6-phosphate specific antibody fragment for diagnosis of Mucolipidosis type II and III (Book chapter) Pohl S, Braulke T, Müller-Loennies S (2012) In: Anticarbohydrate antibodies - From molecular basis to clinical application (Eds: P. Kosma, S. Müller-Loennies), Springer-Verlag, Wien
Lysosomal dysfunction causes neurodegeneration in mucolipidosis II ‘knock-in’ mice Kollmann K, Damme M, Markmann S, Morelle W, Schweizer M, Hermans-Borgmeyer I, Röchert AK, Pohl S, Lübke T, Michalski JC, Käkelä R. Walkley SU, Braulke T (2012) Brain 135: 2661-2675
Multiple Enzyme Deficiencies: Defects in transport: Mucolipidosis II alpha/beta; mucolipidosis III alpha/beta and mucolipidosis III gamma (Book chapter) Raas-Rothschild A, Pohl S, Braulke T (2012) In: Lysosomal Storage Diseases: A Practical Guide (Eds. A. B. Mehta, B. Winchester), Wiley-Blackwell, Oxford
2011
A key enzyme in the biogenesis of lysosomes is a protease that regulates cholesterol metabolism
Marschner K, Kollmann K, Schweizer M, Braulke T, Pohl S (2011) Science 333: 87-90Analysis of potential biomarkers and modifier genes affecting the clinical course of CLN3 disease
Lebrun AH, Moll-Khosrawi P, Pohl S, Makrypidi G, Storch S, Kilian D, Streichert T, Otto B, Mole SE, Ullrich K, Cotman S, Kohlschütter A, Braulke T, Schulz A (2011) ) Mol Med 17: 1253-1261Residual activity and proteasomal degradation of p.Ser298Pro sulfamidase identified in patients with a mild clinical phenotype of Sanfilippo A syndrome
Muschol N, Pohl S, Meyer A, Gal A, Ullrich K, Braulke T (2011) Am J Med Genet A 155: 1634-1639 AbstractPost-translational modifications of the gamma-subunit affect intracellular trafficking and complex assembly of GlcNAc-1-phosphotransferase
Encarnação M, Kollmann K, Trusch M, Braulke T, Pohl S (2011) J Biol Chem 286: 5311-53182010
Proteolytic processing of the gamma-subunit is associated with the failure to form GlcNAc-1-phosphotransferase complexes and mannose 6-phosphate residues on lysosomal enzymes in human macrophages
Pohl S, Tiede S, Marschner K, Encarnação M, Castrichini M, Kollmann K, Muschol N, Ullrich K, Müller-Loennie S, Braulke T (2010) J Biol Chem 285: 23936-23944Mannose phosphorylation in health and disease (Review)
Kollmann K, Pohl S, Marschner K, Encarnação M, Sakwa I, Tiede S, Poorthuis BJ, Lübke T, Müller-Loennies S, Storch S, Braulke T (2010) Eur J Cell Biol 89: 117-123Loss of N-acetylglucosamine-1-phosphotransferase gamma-subunit due to intronic mutation in GNPTG causes mucolipidosis type III gamma: Implications for molecular and cellular diagnostics
Pohl S, Encarnacão M, Castrichini M, Müller-Loennies S, Muschol N, Braulke T (2010) Am J Med Genet A 152: 124-1322009
Compensatory expression of human N-acetylglucosaminyl-1 phosphotransferase subunits in mucolipidosis type III gamma
Pohl S, Tiede S, Castrichini M, Cantz M, Gieselmann V, Braulke T (2009) Biochim Biophys Acta 1792: 221-225Glycosylation- and phosphorylation-dependent intracellular transport of lysosomal hydrolases (Review)
Pohl S, Marschner K, Storch S, Braulke T (2009) Biol Chem 390:521-7Identification and molecular characterization of six novel mutations in the UDP-N-acetylglucosamine-1-phosphotransferase gamma subunit (GNPTG) gene in patients with mucolipidosis III gamma
Persichetti E, Chuzhanova NA, Dardis A, Tappino B, Pohl S, Thomas NS, Rosano C, Balducci C, Paciotti S, Dominissini S, Montalvo AL, Sibilio M, Parini R, Rigoldi M, Di Rocco M, Parenti G, Orlacchio A, Bembi B, Cooper DN, Filocamo M, Beccari T (2009) Hum Mutat 30: 978-9842008
Molecular analysis of the GlcNac-1-phosphotransferase (Review) Braulke T, Pohl S, Storch S (2008) J Inherit Metab Dis 31: 253-257
2007
C-terminal prenylation of the CLN3 membrane glycoprotein is required for efficient endosomal sorting to lysosomes
Storch S, Pohl S, Quitsch A, Falley K, Braulke T (2007) Traffic 8: 431-44Increased expression of lysosomal acid phosphatase (LAP/Acp2) in CLN3-defective cells and mouse brain tissue
Pohl S, Mitchison HM, Kohlschütter A, van Diggelen, O, Braulke T, Storch S (2007) J Neurochem 103: 2177-21882004
A dileucine motif and a cluster of acidic amino acids in the second cytoplasmic domain of the Batten disease-related CLN3 Protein are required for efficient lysosomal targeting
Storch S, Pohl S, Braulke T (2004) J Biol Chem 279: 53625-53634 -


-
Förderung
Laufende Förderung

2024-2028
Deutsche Forschungsgemeinschaft
Klinische Forschungsgruppe KFO 5029:
"ProBone, Präzisionsmedizin bei Erkrankungen mit früh-manifester Reduktion der Knochenmineraldichte"
2023 - 2024
Deutsche Forschungsgemeinschaft
Sachbeihilfe „Die molekulare Rolle der Site-2-Protease für die Funktion der Osteoblasten“
2023-2025
Werner Otto Stiftung
"Verschiedene Rollen der Site-1- und Site-2-Proteasen in der Pathogenese von seltenen, genetisch bedingten Bindegewebeerkrankungen im Kindesalter"
2022 - 2026
Deutsche Forschungsgemeinschaft
Heisenberg-Förderung „Pathophysiologie und molekulare Ursachen bei skelettalen Erkrankungen im Kindesalter“
2018 - 2025
Deutsche Forschungsgemeinschaft
Sachbeihilfe „Rolle der gamma-Untereinheit des GlcNAc-1-Phosphotransferase-Komplexes für selektive Mannose-6-Phosphat-Modifikationen an lysosomalen Enzymen“Abgeschlossene Förderung

2010 - 2022
Deutsche Forschungsgemeinschaft
Sonderforschungsbereich 877:
"Proteolyse als regulatorisches Ereignis in der Pathophysiologie"
www.uni-kiel.de/Biochemie/sfb877/
2017 - 2018
ISMRD - International Society for Mannosidosis & Related Diseases (USA)
"Osteoporosis in Mucolipidosis II - A potential corrective approach"
http://www.ismrd.org/research
2012 - 2017
Deutsche Forschungsgemeinschaft
Graduiertenkolleg 1459:
"Sortierung und Wechselwirkung zwischen Proteinen subzellulärer Kompartimente" -
-
Stellenangebote
Bachelor- und Masterstudierende sind ein integraler Bestandteil unseres Forschungsteams. Wir bieten Forschungsprojekte für Bachelor- und Masterarbeiten an, die sich mit den Pathomechanismen von Skeletterkrankungen befassen.
Zur Unterstützung unserer Forschung suchen wir außerdem Studentische Hilfskräfte.
Wir freuen uns immer über exzellente Doktorand:innen und Postdoktorand:innen, die sich für unsere Forschung interessieren und bereit sind, sich um ein Stipendium zu bewerben, um in unserem Labor mitzuarbeiten.
Wir laden auch Medizinstudierende des UKE ein, ihre Studienarbeit über Knochen- und/oder Knorpelerkrankungen sowie die molekularen Pathomechanismen dieser Erkrankungen und die aktuellen therapeutischen Strategien zu schreiben.
Bitte senden Sie Ihre Anfragen an Prof. Dr. Sandra Pohl (s.pohl@uke.de).